分子生成(一) REINVENT:使用强化学习生成模型产生新的分子
在药物设计、化学信息学领域,深度学习并不一定能比传统机器学习领域有更好的表现,尤其是在学术界,由于有标签的数据很少,数据噪音较大,往往用深度学习只会导致过拟合降低模型的预测能力。但是,生成学习用于药物设计却完全发挥了当前深度神经网络的优势。虽然有标签的数据很少,但是药物设计数据库诸如CHEMBL不乏质量较高的“化合物信息”。这里说的质量较高指的是他们的类药性较强,往往此类数据库中的化合物都是早期药物设计中已经合成出且具有一定生物活性的分子。这些化合物可以统统拿来构建一个模型用于生成新的小分子。
另一方面,深度学习所推崇的”原始数据”,对于图形数据,序列数据的处理能力远远大于传统的机器学习方法,从而解决了传统方法用分子描述符和分子指纹描述分子并不能得到分子结构本身。现在有了深度学习,一切从分子本身(比如分子的 SMILES)进行训练,生成新的SMILE即得到小分子的结构。
RNN生成模型
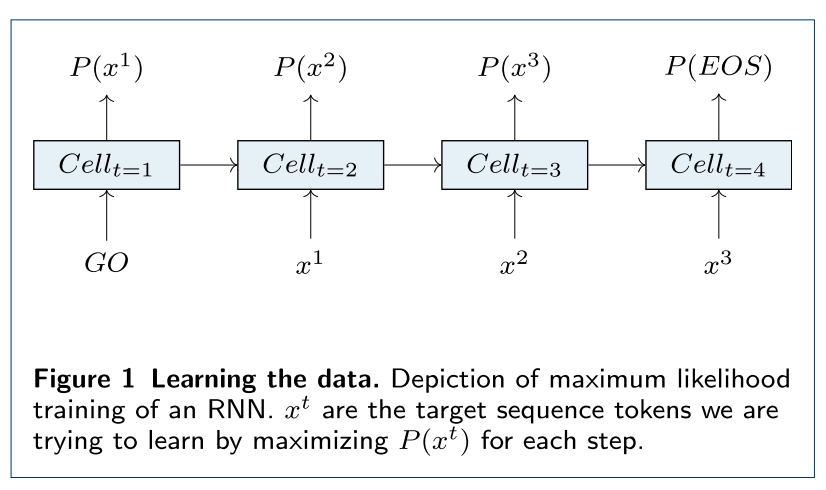
生成模型的架构主要是循环神经网络 (RNN),从下图可以看出,在一个 RNN
模型里,最初的输入是“GO”,接下来每个输入都是上个单元期待的输出,最终输出是“EOS”。比如一个乙醇分子
CCO,对应这里的 x1, x2, x3。训练的目的就是让x1=C,
x2=C,
x3=O的概率之积最高,用损失函数表示就是:
\[J(\Theta)=-\sum_{t=1}^T \log P\left(X^t \mid x^{t-1}, \ldots, x^1\right)\]
中间的竖线是条件符号,表示给定前面的变量后,求后面的变量的概率。在这个公式中,它表示给定 \(x^{t-1}, \ldots, x^1\) 后,求 \(X^t\) 的概率。
取对数之后,连乘变为连加(\(LogMN=LogM+LogN\)),由于
所谓\(Log\),其实就是已知底数和y值求指数。
加上负号是为了将最大化问题转化为最小化问题。
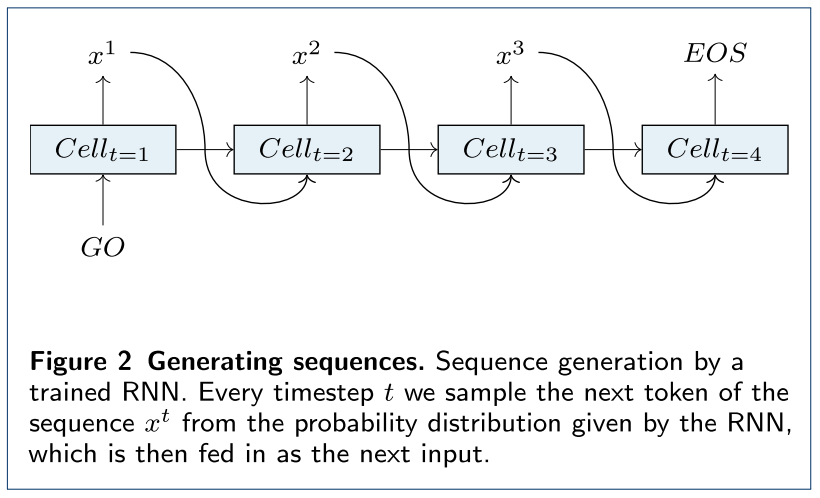
当需要产生新的分子时,则只要让输入时“GO”,然后让前一个单元的输出作为后一个单元的输入,直到有一个单元输出“EOS”表示完成一条数据的生成。
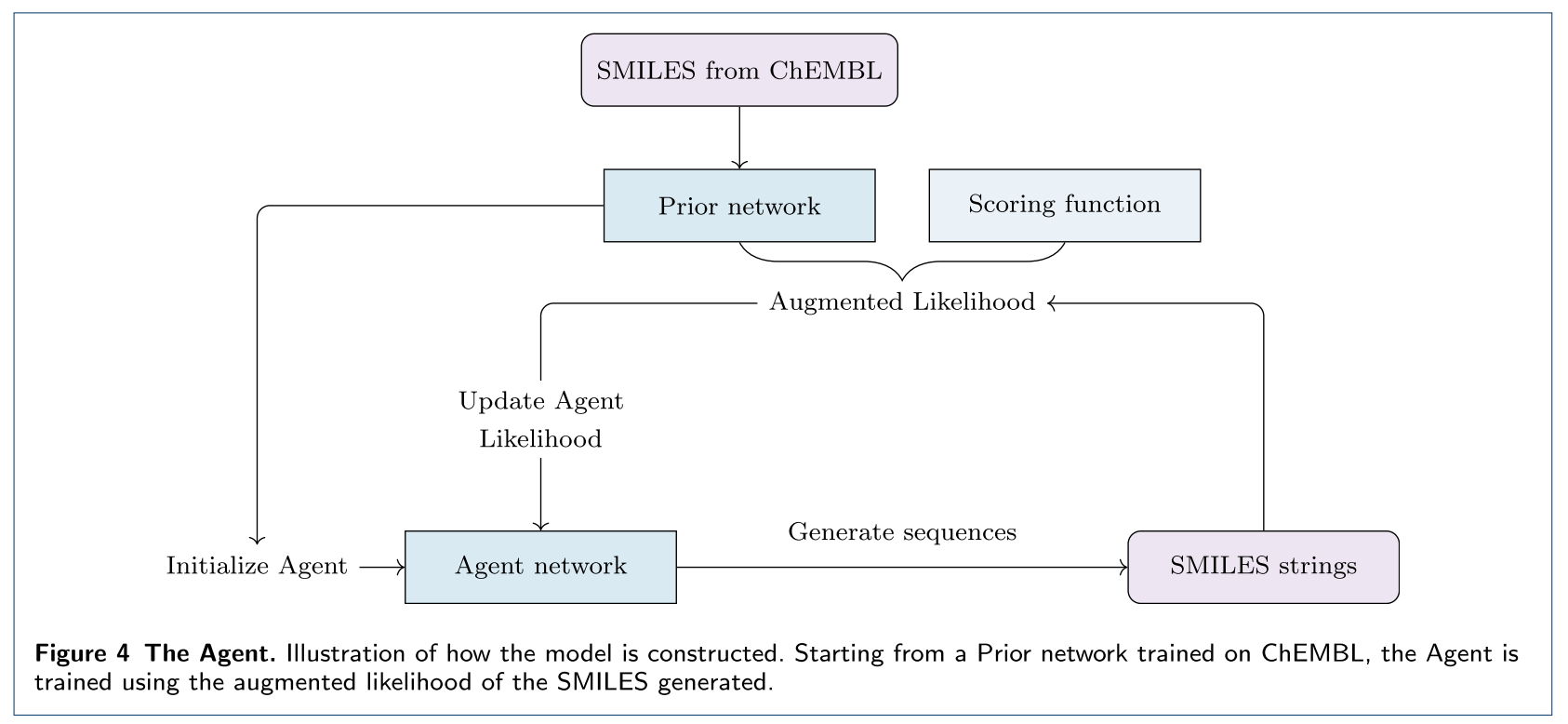
RL优化生成的化合物
当使用大量数据训练RNN生成模型后,该模型(称之为 Prior)具有生成 SMILES 的能力,接下来是通过强化学习来优化这个模型(Agent),让其能生成“更好的分子”。
时可以将生成小分子的过程想象成一场游戏,每生成一个字母就是一个行为(action, \(A={a_1,a_2,..a_n}\)),生成完整个小分子就是完成了整场游戏(episode),获得一个“分数”(\(S(A)\))【-1,-1】。优化的目的是尽可能让生成的小分子拥有较高的分数。比如这里的分数可以是“与某个分子的相似程”。 于是得到一个小分子的概率就可以描述成:
\[P(A)=\prod_{t=1}^T \pi\left(a_t \mid s_t\right)\]
在\(s_t\)状态下执行\(a_t\)的奖励最大化
而具体损失函数如下得到: 首先我们要在Prior模型的基础上增加打分,成为“目标概率”,即:
\[\log P(A)_U=\log P(A)_{\text {Prior }}+\sigma S(A)\]
损失函数则要让Agent做这个决策的概率接近这个目标概率,即:
\[L(\Theta)=\left[\log P(A)_U-\log P(A)_A\right]^2\]
这里希望Agent和Prior保持类似的分布。
实验结果
- 两种模型生成的非含硫结构的模型性能和性能比较

相似化合物生成

DRA2 靶点 活性化合物生成

源码及使用方法
源码和使用方法见:https://github.com/MarcusOlivecrona/REINVENT